The Science Behind an Upcoming Treatment for LCA10/LCA-CEP290

Read this article in Dutch.
Read this article in French.
Dr. Rob Collin at the Radboud University Medical Center in Nijmegen, the Netherlands, is a known expert in the field of retinopathies. At the end of April 2016, my son Enzo and I visited Rob’s research group “Blindness Genetic Therapy” (figure 1).

Figure 1: Photo showing Rob Collin and his 8 group members.
His last work had just been published and thus he enthusiastically guided me through his last masterpiece. At the same time he gave me some background information on the specific CEP290 mutation that he is working on and his collaboration with ProQR, the pharmaceutical company that is about to start the clinical trial. But, first a short summary on basic genetics important for Rob’s work.
The basics
First of all, human cells need two sets of genes to function properly, meaning that we have two copies of each gene. We all have received one set of genes from our mother and one set of genes from of our father. Secondly, our cells can transcribe genes in our DNA into an intermediate (messenger RNA) which is then translated into protein. Every gene contains information that is used to create the later protein (exons) and information that is guiding the building of this protein (introns). Within the exons specific combinations of building blocks, called letters or nucleotides, tell us that transcription should start or stop, the section between this start and stop position is called the coding region.
Let’s now look at the CEP290 mutation that Rob Collin is working on and see if we understand where all this jargon comes in (also see figure 2). The CEP290 gene consists of 54 exons and the most recurrent mutation, c.[2991+1655A>G], is positioned in the intron between exon 26 and 27. Codon (c) position 2991 is the end of exon 26 and now we have to go 1655 letters into the coming intron to find the letter that is mutated from an A (adenine) to a G (guanine). This one letter change results in the insertion of an aberrant pseudo exon (X) with a new stop signal resulting in an incorrect protein and fewer and shorter cilia on the retinal cells.
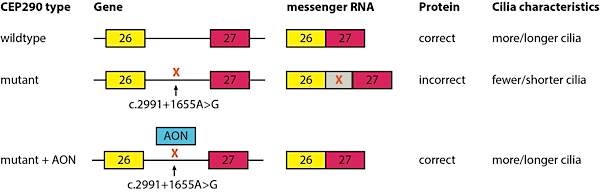
Figure 2: The most recurrent CEP290 mutation, c.[2991+1655A>G], is positioned in the intron between exon 26 and 27. This mutation creates the incorporation of a pseudo exon (X) into the messenger RNA resulting in an incorrect protein and fewer and shorter cilia on cells.
Since we all have two copies of each gene we will only experience vision problems when both copies of the CEP290 gene are affected by mutations, however, these mutations are not necessarily the same. To read more about cilia function and the role of CEP290 in this process you can go to my previous blog post, CEP290 in LCA and other Ciliopathies.
What should readers understand about the treatment that you are developing?
The genetic therapy approach that we have been developing is based on antisense-oligonucleotides (AON). These are small molecules that will mask the pseudo exon ensuring the translation of a correct CEP290 protein. We have tested the effect of AONs on skin cells from LCA-CEP290 (c.[2991+1655A>G]) patients and observed that mRNA type, protein levels and ciliary characteristics were all normalized.
How do you plan to administer this treatment?
AONs can be delivered as naked molecules or packaged in adeno-associated viruses (AAV) which require different administration routes. AAVs are quite large and do not diffuse so far. Thus, they need to be injected close to the retinal cells. ProQR is developing a naked molecule, which is smaller and diffuses more easily and thus can be injected further away directly in the eye, specifically in the vitreous, the jelly-like substance that fills the back of the eye. The administration of naked molecules to the vitreous is a less invasive procedure and therefore more convenient for treatments that are not a “forever fix.” Moreover, we already know from anti-VEGF treatments for macular degeneration that it is feasible to perform repeated injections.
What exciting new things are you trying currently in your lab?
Our previous gene treatment research has all been done on skin cells from patients that have LCA because of two identical mutations in CEP290; they carry two times c.[2991+1655A>G]. However, many patients carry this mutation once and carry another CEP290 mutation as well. We think that those patients would also benefit from the treatment that we are developing, in a sense they should then resemble carriers, eg. as mothers and fathers. Therefore, we are now investigating the cilia structure of skin cells (note: all human cells have a cilium) from carriers allowing us to know what to look for when trying the treatment on skin cell from patients with two different CEP290 mutations.
You are collaborating with ProQR to get this treatment on the market. Do you have any idea about the timing?
There is still a long way to go before this potential treatment will be available to patients. We want to start the first clinical trial as soon as possible. However, preparations are still ongoing and there is no precise timing for the start of the study yet.
Sources:
- https://exonskipping.eu/participant/rob-collin/
- https://www.proqr.com/lca/
- Garanto A, Chung DC, Duijkers L et al., In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum Mol Genet. 2016 Apr 22.
About the author : My name is Laura Steinbusch and together with Merlijn and Enzo I live in Lausanne, Zwitserland. Enzo was born in August 2014 and he is blind because of Leber’s Congenital Amaurosis (LCA).

The information WonderBaby provides is not intended to be, and does not constitute, medical or other health advice or diagnosis and should not be used as such. Always consult with a qualified medical professional about your specific circumstances.
Related Posts

Eye Conditions and Syndromes, Visual Impairment
Neuralink Announces Plans to Restore Sight to the Blind with Brain Chip
Elon Musk’s company Neuralink has announced plans to begin human trials of its new “Blindsight” brain chip by the end of 2025.

Eye Conditions and Syndromes
Does Screen Time Affect Kids’ Vision?
Too much screen time can affect kids’ vision by causing eye strain, blurred vision, dry eyes, and even nearsightedness in children and adolescents.

Eye Conditions and Syndromes, Support, Visual Impairment
Coping with a Diagnosis: Emotional Support for Families with Visually Impaired Children
Families with emotional support are more resilient. Learn how to establish emotional support with peers, professionals, and the community to help your family thrive.